Site-Specific Inhibition of Transcription Factor Binding
to DNA by a Metallointercalator![]()
Web Release Date: April 1,
Site-Specific Inhibition of Transcription Factor Binding
to DNA by a Metallointercalator![]()
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125
Received November 25, 1998
Revised Manuscript Received February 3, 1999
Abstract:
The metallointercalator ![]() -1-Rh(MGP)2phi5+
binds tightly and specifically to the site 5'-CATATG-3' in the major groove of
double helical DNA by a combination of direct readout and shape selection. To
examine competitive interactions between this small metal complex and a
DNA-binding transcription factor, the preferred binding site for
-1-Rh(MGP)2phi5+
binds tightly and specifically to the site 5'-CATATG-3' in the major groove of
double helical DNA by a combination of direct readout and shape selection. To
examine competitive interactions between this small metal complex and a
DNA-binding transcription factor, the preferred binding site for ![]() -1-Rh(MGP)2phi5+
was engineered into the AP-1 recognition element (ARE) of the major-groove
binding bZIP transcription factor yAP-1, the yeast analogue of mammalian AP-1.
Binding experiments confirmed that the modified ARE retained normal yAP-1
binding affinity. Photocleavage experiments demonstrated that the modified ARE
contained a high-affinity binding site for
-1-Rh(MGP)2phi5+
was engineered into the AP-1 recognition element (ARE) of the major-groove
binding bZIP transcription factor yAP-1, the yeast analogue of mammalian AP-1.
Binding experiments confirmed that the modified ARE retained normal yAP-1
binding affinity. Photocleavage experiments demonstrated that the modified ARE
contained a high-affinity binding site for ![]() -1-Rh(MGP)2phi5+,
whereas the native ARE showed no interaction. Competition experiments using gel
shift mobility assays demonstrated that
-1-Rh(MGP)2phi5+,
whereas the native ARE showed no interaction. Competition experiments using gel
shift mobility assays demonstrated that ![]() -1-Rh(MGP)2phi5+
at 120 nM competes 50% of yAP-1 binding to the 5'-CATATG-3' containing
oligonucleotide. In contrast, competitive disruption of protein binding to the
native ARE requires 3
-1-Rh(MGP)2phi5+
at 120 nM competes 50% of yAP-1 binding to the 5'-CATATG-3' containing
oligonucleotide. In contrast, competitive disruption of protein binding to the
native ARE requires 3 ![]() M
M ![]() -1-Rh(MGP)2phi5+.
Metallointercalator derivatives, including geometric isomers of
-1-Rh(MGP)2phi5+.
Metallointercalator derivatives, including geometric isomers of ![]() -1-Rh(MGP)2phi5+,
show no specific binding to the target site and show no inhibition of yAP-1/DNA
complexes at concentrations as high as 20
-1-Rh(MGP)2phi5+,
show no specific binding to the target site and show no inhibition of yAP-1/DNA
complexes at concentrations as high as 20 ![]() M.
Thus, metallointercalators can be tuned to show selectivity for major groove
sites on DNA comparable to transcription factors and indeed can inhibit
transcription factor binding site selectively.
M.
Thus, metallointercalators can be tuned to show selectivity for major groove
sites on DNA comparable to transcription factors and indeed can inhibit
transcription factor binding site selectively.
Proteins that interact with DNA are the subjects of great interest in
developing potential targets for therapeutic treatment. Since transcription
factors modulate the expression of genes by binding to and interacting with
specific DNA sequences, one frequent target for drug design and biochemical
modification has been the DNA-binding sites of transcription factors. One method
for altering transcription regulation both in vivo and in vitro is to modify the
DNA bases of the protein binding site. Enzymatic base methylation (1),
the formation of cisplatin lesions (2-4)
Alternatively, agents which bind DNA noncovalently may be used to interfere
competitively with DNA-binding proteins. One method for targeting specific
genetic loci has been to use complementary protein nucleic acids (PNAs);
however, these molecules may lack the cell permeability that is needed to make
them effective therapeutic agents (7). The
natural product calicheamicin as well as some derivatives have been shown to
bind sequence selectively to the minor groove of DNA and to inhibit
transcription factor binding (8-10)
Another particularly promising class of potential therapeutics is the polyamides. These small synthetic molecules bind to DNA in the minor groove with high sequence specificity (11) and have been shown to inhibit transcription both in vitro and in vivo (12). Often in conjunction with major groove contacts, the minor groove is contacted by many transcription factors. By disrupting the minor groove contacts, a number of transcription factors can be displaced.
Currently, however, few strategies exist that utilize the major groove directly as a surface for recognition by small molecules to inhibit the initiation of transcription. Because the major groove is preferentially recognized by transcription factors (7), small molecules that recognize specified regions within the major groove might be especially effective in blocking DNA binding. The steric accessibility of the wide and deep major groove, the greater range of functionality within the major groove versus the minor groove, and the possibility of cooperative, multiple protein-DNA contacts in the major groove make it an attractive target for therapeutic design. Hence, it would be of great interest to find a class of small molecules that can competitively inhibit the binding of transcription factors in the major groove.
Our research has focused on the development of metallointercalators that bind
to DNA in the major groove (13, 14)![]() -
-![]() -[Rh[(R,R)-Me2trien]phi]3+
(Me2trien=2,9-diamino-4,7-diazadecane) are octahedral complexes which
are coordinatively saturated and inert to ligand substitution. The phi ligand,
an extended aromatic, heterocyclic surface, preferentially intercalates between
the DNA base pairs, and in so doing, the intercalated phi orients the rigid
complex with respect to the helix. All phi complexes of rhodium examined thus
far appear to intercalate into the major groove side of the DNA duplex (13-17)
-[Rh[(R,R)-Me2trien]phi]3+
(Me2trien=2,9-diamino-4,7-diazadecane) are octahedral complexes which
are coordinatively saturated and inert to ligand substitution. The phi ligand,
an extended aromatic, heterocyclic surface, preferentially intercalates between
the DNA base pairs, and in so doing, the intercalated phi orients the rigid
complex with respect to the helix. All phi complexes of rhodium examined thus
far appear to intercalate into the major groove side of the DNA duplex (13-17)![]() -
-![]() -[Rh[(R,R)-Me2trien]phi]3+.
This complex recognizes its target site 5'-TGCA-3' through a mixture of
hydrogen-bonding interactions between the axial amines of the complex and
guanine bases and van der Waals contacts between methyl groups on the ligands
and on the thymine residues positioned in the major groove (15,
18)
-[Rh[(R,R)-Me2trien]phi]3+.
This complex recognizes its target site 5'-TGCA-3' through a mixture of
hydrogen-bonding interactions between the axial amines of the complex and
guanine bases and van der Waals contacts between methyl groups on the ligands
and on the thymine residues positioned in the major groove (15,
18)
Here, our focus is on phi complexes of rhodium that have functionalized
phenanthrolines as ancillary ligands (Figure 1). These complexes have been shown
to exploit a combination of direct readout and shape selectivity to recognize
sites in DNA (13, 20)
 |
Figure 1 |
The pendant guanidiniums on the ancillary phenanthroline rings of Rh(MGP)2phi5+ (MGP = 4-guanidylmethyl-1,10-phenanthroline) lead instead to a high level of DNA site specificity (Figure 1). The guanidinium groups of arginine side chains are exploited frequently by proteins in targeting guanine residues of DNA (22). Because the MGP ligand is asymmetrically functionalized, multiple regioisomers of the complex exist. There are three geometric isomers, each of which has two stereoisomers. Remarkably, each of these different isomers exhibits quite different recognition of DNA, demonstrating that the metal complex binding is dependent upon the alignment of functional groups. Notably, the symmetric isomer 2, with guanidiniums pointing away from the phi ligand, shows sequence selectivity resembling that of Rh(phen)2phi3+.
Of particular interest, the symmetric isomer ![]() -1-Rh(MGP)2phi5+,
with guanidinium arms that extend axially from the intercalating phi plane
forward over the phi ligand, displays strong and site-selective binding to DNA.
-1-Rh(MGP)2phi5+,
with guanidinium arms that extend axially from the intercalating phi plane
forward over the phi ligand, displays strong and site-selective binding to DNA. ![]() -1-Rh(MGP)2phi5+
binds preferentially to the site 5'-CATATG-3' with nanomolar affinity. Although
there is some tolerance for differences at the outermost positions in the 6 base
pair site (13), any variation of the
central 5'-ATAT-3' disfavors metal complex intercalation. Molecular modeling
shows that, in the absence of DNA distortion, the stereochemistry of the
guanidinium groups on the complex does not permit the bases at the extremities
to be contacted by the guanidiniums, yet binding experiments in which guanines
were replaced with deazaguanine have shown that terminal guanine-guanidinium
contacts occur. DNA cyclization assays established that the intercalation of the
metal complex is associated with a 70
-1-Rh(MGP)2phi5+
binds preferentially to the site 5'-CATATG-3' with nanomolar affinity. Although
there is some tolerance for differences at the outermost positions in the 6 base
pair site (13), any variation of the
central 5'-ATAT-3' disfavors metal complex intercalation. Molecular modeling
shows that, in the absence of DNA distortion, the stereochemistry of the
guanidinium groups on the complex does not permit the bases at the extremities
to be contacted by the guanidiniums, yet binding experiments in which guanines
were replaced with deazaguanine have shown that terminal guanine-guanidinium
contacts occur. DNA cyclization assays established that the intercalation of the
metal complex is associated with a 70![]() unwinding at the DNA site, and this unwinding explains how the positioning of
guanine-guanidinium contacts can occur. NMR evidence further suggests that the
metal complex traps the DNA site in an unwound form, hence the strict
requirement for a central TA-rich stretch (23).
unwinding at the DNA site, and this unwinding explains how the positioning of
guanine-guanidinium contacts can occur. NMR evidence further suggests that the
metal complex traps the DNA site in an unwound form, hence the strict
requirement for a central TA-rich stretch (23).
The six base pair recognition of a DNA site by this small synthetic complex,
as well as the unwinding and exchange kinetics associated with binding, may
resemble the interactions of many proteins that bind to DNA site specifically.
Significant bend angles are often found in DNA remodeling proteins, for
instance, TATA box binding protein (TBP) (24,
25)![]() -1-Rh(MGP)2phi5+
similar to TBP in that it uses guanidinium functionalities to generate a network
of hydrogen bonds to DNA, but with both TBP and the synthetic metal complex,
significant unwinding (70
-1-Rh(MGP)2phi5+
similar to TBP in that it uses guanidinium functionalities to generate a network
of hydrogen bonds to DNA, but with both TBP and the synthetic metal complex,
significant unwinding (70![]() vs 108
vs 108![]() for TBP) and kinking of the bound site occurs. Indeed, the preferential
recognition of sites by TBP that contain TA-rich stretches may share mechanistic
features with the targeting of
for TBP) and kinking of the bound site occurs. Indeed, the preferential
recognition of sites by TBP that contain TA-rich stretches may share mechanistic
features with the targeting of ![]() -1-Rh(MGP)2phi5+
to its preferred binding site (13).
-1-Rh(MGP)2phi5+
to its preferred binding site (13).
One of the prominent classes of proteins that site specifically interact with
DNA is the bZIP transcription factors (26).
Several of these proteins bound to their target DNA sites have been structurally
characterized, among them is the transcription factor GCN4, and in these
structures, a common structural motif is apparent. Most prominent is the
dimerization domain which includes a coiled coil ![]() -helical
region stabilized by the packing of leucine side chains; the dimerization domain
is capped by a highly basic, DNA-binding region. Separate regions either
upstream or downstream of the bZIP DNA binding motif regulate transcriptional
activation and signaling. Another member of this class of transcription factors
is the yeast analogue of mammalian AP-1, yAP-1, which is 70 kDa in size (27).
yAP-1 shows sequence homology to other bZIP transcription factors, and the
isolated DNA-binding region from yAP-1 shows the induction of an
-helical
region stabilized by the packing of leucine side chains; the dimerization domain
is capped by a highly basic, DNA-binding region. Separate regions either
upstream or downstream of the bZIP DNA binding motif regulate transcriptional
activation and signaling. Another member of this class of transcription factors
is the yeast analogue of mammalian AP-1, yAP-1, which is 70 kDa in size (27).
yAP-1 shows sequence homology to other bZIP transcription factors, and the
isolated DNA-binding region from yAP-1 shows the induction of an ![]() helix upon binding to its DNA recognition site, consistent with the leucine
zipper motif (28). Functionally, yAP-1
regulates pleiotropic drug resistance and has been shown to be nonessential for
cell viability under normal circumstances (29).
yAP-1 has a strong affinity for the site 5'-AATTAGTCAGCA-3', known as the
activator recognition element (ARE). Aspects of how yAP-1 recognizes its target
site have been determined by chemical probing with dimethyl sulfate (DMS), DNAse
I, and methidiumpropylEDTA iron digestion (MPE) (Figure 2, top sequence) (30).
Footprinting with MPE suggests that the yAP-1-binding domain spans the consensus
site, but DMS digestion of DNA in the presence of yAP-1 reveals that there are a
number of base pairs within the core of the consensus region that do not appear
to be in intimate contact with the protein. This apparent lack of contact at
particular locations within the site suggested the target site could be mutated
to contain an overlapping site for
helix upon binding to its DNA recognition site, consistent with the leucine
zipper motif (28). Functionally, yAP-1
regulates pleiotropic drug resistance and has been shown to be nonessential for
cell viability under normal circumstances (29).
yAP-1 has a strong affinity for the site 5'-AATTAGTCAGCA-3', known as the
activator recognition element (ARE). Aspects of how yAP-1 recognizes its target
site have been determined by chemical probing with dimethyl sulfate (DMS), DNAse
I, and methidiumpropylEDTA iron digestion (MPE) (Figure 2, top sequence) (30).
Footprinting with MPE suggests that the yAP-1-binding domain spans the consensus
site, but DMS digestion of DNA in the presence of yAP-1 reveals that there are a
number of base pairs within the core of the consensus region that do not appear
to be in intimate contact with the protein. This apparent lack of contact at
particular locations within the site suggested the target site could be mutated
to contain an overlapping site for ![]() -1-Rh(MGP)2phi5+
without affecting the ARE specific binding of the protein. Such a DNA site would
allow competitive assays to be carried out between yAP-1 and
-1-Rh(MGP)2phi5+
without affecting the ARE specific binding of the protein. Such a DNA site would
allow competitive assays to be carried out between yAP-1 and ![]() -1-Rh(MGP)2phi5+.
-1-Rh(MGP)2phi5+.
 |
Figure 2 Modifications made to the ARE to incorporate a metal complex binding site (adapted from ref 14). The top oligonucleotide sequence (labeled wild-type) is the wild-type ARE, capped with GCG triplets. The boxed regions show protection against MPE footprinting in the presence of yAP-1. The solid triangles indicate increases or decreases in sensitivity in DMS labile sites when footprinted in the presence of yAP-1. The target strand, below, has incorporated a 5'-CATATG-3' site with no disruption of the footprinted nucleotides. The bases modified are shown in bold italics. |
The notion of establishing a functional similarity between ![]() -1-Rh(MGP)2phi5+
and a DNA-binding proteins such as yAP-1 in targeting a specific site is
interesting to consider in light of the much smaller size and relative
simplicity of the transition metal complex. If this metal complex could be used
to inhibit the binding of a protein to DNA by recognition of a common site, this
reactivity would strengthen the possible application of metallointercalators as
competitive agents to alter transcription. The findings reported herein
demonstrate that indeed
-1-Rh(MGP)2phi5+
and a DNA-binding proteins such as yAP-1 in targeting a specific site is
interesting to consider in light of the much smaller size and relative
simplicity of the transition metal complex. If this metal complex could be used
to inhibit the binding of a protein to DNA by recognition of a common site, this
reactivity would strengthen the possible application of metallointercalators as
competitive agents to alter transcription. The findings reported herein
demonstrate that indeed ![]() -1-Rh(MGP)2phi5+
can be used to inhibit site specifically the binding of a transcription factor
to a promoter site in vitro. We show that this interaction is dependent both on
the identity of the metal complex and on the presence of a metal complex binding
site.
-1-Rh(MGP)2phi5+
can be used to inhibit site specifically the binding of a transcription factor
to a promoter site in vitro. We show that this interaction is dependent both on
the identity of the metal complex and on the presence of a metal complex binding
site.
All chemicals and biochemicals were from commercial sources. Beckman JA2-21
and JA-4 centrifuges were used for general purposes. A Branson Sonifier 450 was
used to lyse cell preparations. All enzymes were purchased from commercial
sources unless otherwise noted. All proteins were stored at -20 ![]() C
in protein buffer that consisted of 10% glycerol, 25 mM hepes, pH 7.4, 1 mM EDTA,
50 mM KCl, and 0.1% Nonidet-40.
C
in protein buffer that consisted of 10% glycerol, 25 mM hepes, pH 7.4, 1 mM EDTA,
50 mM KCl, and 0.1% Nonidet-40.
Synthesis and Purification of DNA and Metal Complexes. All DNA was
synthesized with a terminal trityl group on an ABI 392 DNA/RNA synthesizer with
reagents from Glen Research. HPLC purification and subsequent detritylation used
standard techniques. All labeled oligonucleotides were 5'-end labeled using
standard protocols with polynucleotide kinase (New England Biolabs) and ![]() -32P-ATP.
The labeled DNA was purified by denaturing gel electrophoresis on a 20%
acrylamide gel, followed by crush and soak elution in 10 mM Tris-HCl, pH 7.4,
and 1 mM EDTA. The purified labeled material was then annealed to a 10-fold
excess of complement, and native gel electrophoresis performed on a 10%
polyacrylamide gel containing 90 mM Tris borate, pH 8.3, and 1 mM EDTA. The
synthesis and purification of the metal complexes followed previously reported
procedures (20, 31)
-32P-ATP.
The labeled DNA was purified by denaturing gel electrophoresis on a 20%
acrylamide gel, followed by crush and soak elution in 10 mM Tris-HCl, pH 7.4,
and 1 mM EDTA. The purified labeled material was then annealed to a 10-fold
excess of complement, and native gel electrophoresis performed on a 10%
polyacrylamide gel containing 90 mM Tris borate, pH 8.3, and 1 mM EDTA. The
synthesis and purification of the metal complexes followed previously reported
procedures (20, 31)
Preparation of Recombinant yAP-1 Expression Vector. The plasmid pUC19
containing the complete open reading frame of the YAP1 gene in 2.5 kB of
yeast cDNA was digested with EcoRI, and the YAP1 gene was isolated
by agarose gel electrophoresis. pBluescript IISK(-) was digested with EcoRI
and calf alkaline phosphatase, and gel purified. The previously isolated YAP1
gene was ligated into this pBS vector, and successful ligations were selected by
ampicillin resistance in DH5![]() E. Coli cells. Plasmids from this step were harvested and inserted
into CJ236 (dut-, ung-) cells. K07 helper phage and kanamycin
selection was used with this construction to generate a ssDNA template for
site-directed mutagenesis (32). A 3' SalI
restriction site and a 5' NsiI restriction site were introduced into the YAP1
gene. The gene was excised, isolated, and ligated into a thioredoxin fusion
expression vector pThioHisA (Invitrogen). The 5' and 3' termini of
pThioHis(yAP-1) were sequenced, verifying proper framing and mutagenesis.
E. Coli cells. Plasmids from this step were harvested and inserted
into CJ236 (dut-, ung-) cells. K07 helper phage and kanamycin
selection was used with this construction to generate a ssDNA template for
site-directed mutagenesis (32). A 3' SalI
restriction site and a 5' NsiI restriction site were introduced into the YAP1
gene. The gene was excised, isolated, and ligated into a thioredoxin fusion
expression vector pThioHisA (Invitrogen). The 5' and 3' termini of
pThioHis(yAP-1) were sequenced, verifying proper framing and mutagenesis.
Expression and Purification of Recombinant yAP-1 Protein. The vector
pThioHis(yAP-1) was transfected into DE3 cells (BL21), and four to five colonies
were selected from an LB-ampicillin plate incubated overnight at 37 ![]() C.
These colonies were used directly to infect 1 L of LB media with 40
C.
These colonies were used directly to infect 1 L of LB media with 40 ![]() g/mL
ampicillin with shaking at 200 rpm at 37
g/mL
ampicillin with shaking at 200 rpm at 37 ![]() C.
At OD595 = 0.6, 10 mL of 100 mM isopropylthiogalactoside was added to
a final concentration of 1 mM, and the cells were transferred to a 30
C.
At OD595 = 0.6, 10 mL of 100 mM isopropylthiogalactoside was added to
a final concentration of 1 mM, and the cells were transferred to a 30 ![]() C
shaker. After 3 h of induction, the cells were harvested by 15 min
centrifugation at 1000g, and frozen overnight at -80
C
shaker. After 3 h of induction, the cells were harvested by 15 min
centrifugation at 1000g, and frozen overnight at -80 ![]() C.
The cell pellet was rinsed with and then resuspended into 20 mL of protein
buffer with 1 mM
C.
The cell pellet was rinsed with and then resuspended into 20 mL of protein
buffer with 1 mM ![]() -toluenesulfonyl
fluoride. To this, 40 mg lysozyme was added, and the solution was incubated at
23
-toluenesulfonyl
fluoride. To this, 40 mg lysozyme was added, and the solution was incubated at
23 ![]() C for 20 min. This solution was then
sonicated at 50% duty cycle for 2 min on ice, followed by immediate 12000g,
10 min centrifugation of cellular debris. The pellet was discarded, and the
lysate was treated with 10% polyethyleneimine to a final concentration of 0.5%.
After 20 min of room-temperature incubation, the precipitated nucleic acids were
spun down at 35000g for 25 min at 0
C for 20 min. This solution was then
sonicated at 50% duty cycle for 2 min on ice, followed by immediate 12000g,
10 min centrifugation of cellular debris. The pellet was discarded, and the
lysate was treated with 10% polyethyleneimine to a final concentration of 0.5%.
After 20 min of room-temperature incubation, the precipitated nucleic acids were
spun down at 35000g for 25 min at 0 ![]() C.
Ammonium sulfate was then added to a concentration of 1.0 M and the solution was
placed on ice for 30 min. The protein pellet was obtained by centrifugation at
35000g for 20 min and was redissolved into 10 mL of protein buffer at 0
C.
Ammonium sulfate was then added to a concentration of 1.0 M and the solution was
placed on ice for 30 min. The protein pellet was obtained by centrifugation at
35000g for 20 min and was redissolved into 10 mL of protein buffer at 0 ![]() C.
All remaining steps were performed at 4
C.
All remaining steps were performed at 4 ![]() C.
After filtration through a 0.22
C.
After filtration through a 0.22 ![]() M filter,
this material was loaded onto a 5 mL Heparin column (Pharmacia), thoroughly
rinsed with 100 mL of protein buffer. yAP-1 eluted at 700 mM NaCl on a
continuous, linear 200 mL 0.01 to 2.00 M NaCl gradient with protein buffer as
the FPLC carry buffer. The resultant material was >90% pure as visualized by
gel electrophoresis and Coomasie Blue staining. This band was positive to
Western antibody probes to both yAP-1 and thioredoxin, and highly active in
DNA/protein gel retardation assays.
M filter,
this material was loaded onto a 5 mL Heparin column (Pharmacia), thoroughly
rinsed with 100 mL of protein buffer. yAP-1 eluted at 700 mM NaCl on a
continuous, linear 200 mL 0.01 to 2.00 M NaCl gradient with protein buffer as
the FPLC carry buffer. The resultant material was >90% pure as visualized by
gel electrophoresis and Coomasie Blue staining. This band was positive to
Western antibody probes to both yAP-1 and thioredoxin, and highly active in
DNA/protein gel retardation assays.
 |
Figure 3 Photocleavage of 5'-32P labeled target by |
Photocleavage Reactions. Photocleavage reactions were performed using
5'-end-labeled oligonucleotides purified as described earlier. Cold carrier
duplex was made using HPLC purified oligonucleotides. The photocleavage
reactions contained 50 mM NaCl and 10 mM sodium cacodylate, pH 7.0, with a 10:1
ratio of DNA to rhodium complex. A 5 min preincubation in the dark was followed
by 8 min irradiations at 313 nm. Samples lacking metal complexes were irradiated
for 15 min. Immediately after irradiation, each sample was frozen at -80 ![]() C
and lyophilized. They were resuspended in formamide loading buffer and directly
run on a denaturing polyacrylamide sequencing gel (8 M urea and 20%
polyacrylamide).
C
and lyophilized. They were resuspended in formamide loading buffer and directly
run on a denaturing polyacrylamide sequencing gel (8 M urea and 20%
polyacrylamide).
Competition Experiments to Measure Protein Binding. Metal complexes
were freshly dissolved into doubly distilled, deionized water, and the
concentrations of the stock solutions were determined by measuring absorbance (![]() 360
= 1.94 × 104 M-1 cm-1). Protein concentrations
were established by dilution from a stock dialyzed against protein buffer that
had been quantitated by 280 nm absorbance to have 6.25
360
= 1.94 × 104 M-1 cm-1). Protein concentrations
were established by dilution from a stock dialyzed against protein buffer that
had been quantitated by 280 nm absorbance to have 6.25 ![]() M
protein. Each reaction had final concentrations as follows: 40 nM yAP-1, 2%
Ficoll, 50 mM KCl, 25 nCurie of labeled oligonucleotide (~2 nM final
concentration of duplex DNA), 1
M
protein. Each reaction had final concentrations as follows: 40 nM yAP-1, 2%
Ficoll, 50 mM KCl, 25 nCurie of labeled oligonucleotide (~2 nM final
concentration of duplex DNA), 1 ![]() M bovine
serum albumin as nonspecific carrier, and 15 mM Tris, pH 7.4. The amount of
protein buffer included in the reaction varied among the experiments but never
exceeded 2% of the final volume (v/v). The incubations were equilibrated for 3-6
h and were then loaded directly onto a running 5.5% native acrylamide gel
buffered with 45 mM tris-borate buffer with 1 mM EDTA at pH 8.3. Order of
incubation was varied and found to be unimportant. The electrophoresis was
performed for 1 h at 100 V, after which the gels were vacuum-dried onto Whatman
paper and exposed overnight to a Phosphorimager plate. Results were quantitated
in ImageQuant, and data fitting was performed on Kaleidograph software, using
the equation
M bovine
serum albumin as nonspecific carrier, and 15 mM Tris, pH 7.4. The amount of
protein buffer included in the reaction varied among the experiments but never
exceeded 2% of the final volume (v/v). The incubations were equilibrated for 3-6
h and were then loaded directly onto a running 5.5% native acrylamide gel
buffered with 45 mM tris-borate buffer with 1 mM EDTA at pH 8.3. Order of
incubation was varied and found to be unimportant. The electrophoresis was
performed for 1 h at 100 V, after which the gels were vacuum-dried onto Whatman
paper and exposed overnight to a Phosphorimager plate. Results were quantitated
in ImageQuant, and data fitting was performed on Kaleidograph software, using
the equation ![]() = 1 - (Ka)(RhT)/[1
+ Ka)(RhT)], where Ka is the
association constant, RhT is the rhodium concentration, and
= 1 - (Ka)(RhT)/[1
+ Ka)(RhT)], where Ka is the
association constant, RhT is the rhodium concentration, and ![]() is the fraction DNA bound (derived from equations in ref 33).
is the fraction DNA bound (derived from equations in ref 33).
Purification of Recombinant Transcription Factor yAP-1. Previous
investigations using recombinant yAP-1 relied on the activity of crude bacterial
cell lysates of bacterial cells carrying the gene on a constituitively producing
locus (30). Introduction of the YAP1
gene to the pThioHis vector generated a thioredoxin/yAP-1 fusion protein. This
fusion protein was additionally placed under the lacZ promoter, which
allows controlled induction of protein expression in bacteria. The thioredoxin
domain increased the temperature stability of yAP-1 so that the fusion protein
was stable in protein buffer at 60 ![]() C for
up to 3 h with no discernible effect on DNA binding. Purifications from cell
lysate by polyethylimine precipitation of cellular nucleic acids and a 25%
ammonium sulfate cut yielded crude yAP-1 separation from the bulk of the
cellular proteins. Finally, the use of a Heparin agarose affinity column
resulted in >90% purity. As expected, gel shift assays showed that the
addition of the thioredoxin fusion domain did not interfere with yAP-1 binding
to oligonucleotides. Because of this maintenance of activity, combined with
previous studies demonstrating the generally small perturbations introduced upon
fusion of the thioredoxin domain (40), we
used the fusion protein directly in assays. Additionally, this fusion generated
a second epitope for antibody detection, so that the purification steps could be
monitored with commercially available antibodies.
C for
up to 3 h with no discernible effect on DNA binding. Purifications from cell
lysate by polyethylimine precipitation of cellular nucleic acids and a 25%
ammonium sulfate cut yielded crude yAP-1 separation from the bulk of the
cellular proteins. Finally, the use of a Heparin agarose affinity column
resulted in >90% purity. As expected, gel shift assays showed that the
addition of the thioredoxin fusion domain did not interfere with yAP-1 binding
to oligonucleotides. Because of this maintenance of activity, combined with
previous studies demonstrating the generally small perturbations introduced upon
fusion of the thioredoxin domain (40), we
used the fusion protein directly in assays. Additionally, this fusion generated
a second epitope for antibody detection, so that the purification steps could be
monitored with commercially available antibodies.
Design of Target Oligonucleotide. The contacts yAP-1 makes to the
native AP-1 recognition element (ARE) have been well-characterized biochemically
and are shown along with the target oligonucleotide in Figure 2 (30).
To permit binding of ![]() -1-Rh(MGP)2phi5+
to a site also recognized by yAP-1, a minimum number of nucleotides not directly
contacted by the protein were modified to introduce the recognition site
preferred by the metal complex, 5'-CATATG-3'. The three base pair alterations
needed to introduce 5'-CATATG-3' into the wild-type ARE removed only one AT base
pair from the AT rich tract that dominates half the ARE, and inverted two
others. yAP-1 interacts with both the modified sequence of the target
oligonucleotide and the native sequence with a dissociation constant that is
less than 5 nM (Figure S1, Supporting Information).
-1-Rh(MGP)2phi5+
to a site also recognized by yAP-1, a minimum number of nucleotides not directly
contacted by the protein were modified to introduce the recognition site
preferred by the metal complex, 5'-CATATG-3'. The three base pair alterations
needed to introduce 5'-CATATG-3' into the wild-type ARE removed only one AT base
pair from the AT rich tract that dominates half the ARE, and inverted two
others. yAP-1 interacts with both the modified sequence of the target
oligonucleotide and the native sequence with a dissociation constant that is
less than 5 nM (Figure S1, Supporting Information).
Recognition of Target Oligonucleotide by ![]() -1-Rh(MGP)2phi5+
as Visualized by Photocleavage. Having established strong interaction
between the transcription factor and the target oligonucleotide sequence, DNA
photocleavage studies were used to probe the interaction of the metal complexes
and oligonucleotides used in this study. As in previous studies, the metal
complexes were preequilibrated for 5 min with the oligonucleotide before
irradiation to ensure equilibration (20);
NMR studies indicate, however, that the equilibration is much more rapid (23).
Upon irradiation,
-1-Rh(MGP)2phi5+
as Visualized by Photocleavage. Having established strong interaction
between the transcription factor and the target oligonucleotide sequence, DNA
photocleavage studies were used to probe the interaction of the metal complexes
and oligonucleotides used in this study. As in previous studies, the metal
complexes were preequilibrated for 5 min with the oligonucleotide before
irradiation to ensure equilibration (20);
NMR studies indicate, however, that the equilibration is much more rapid (23).
Upon irradiation, ![]() -1-Rh(MGP)2phi5+
exhibited strong recognition of the site introduced into the transcription
promoter region (Figure 3) as seen through photocleavage. In contrast, very
little cleavage was observed for wild-type ARE below micromolar concentrations
(Figure S2, Supporting Information). As described previously, the cleavage seen
is a strong doublet at the central AT step of the CATATG-binding site (13),
consistent with the symmetric intercalation of the rhodium complex within its
target site, as observed by NMR (23). We
estimate a dissociation constant of 25 ± 10 nM by photocleavage titration at
this doublet as a function of concentration based on curve fitting (see
Materials and Methods). Photocleavage quantum yields for these metal complexes
are quite low (19), yet it is clear that
binding saturation occurs well below 1
-1-Rh(MGP)2phi5+
exhibited strong recognition of the site introduced into the transcription
promoter region (Figure 3) as seen through photocleavage. In contrast, very
little cleavage was observed for wild-type ARE below micromolar concentrations
(Figure S2, Supporting Information). As described previously, the cleavage seen
is a strong doublet at the central AT step of the CATATG-binding site (13),
consistent with the symmetric intercalation of the rhodium complex within its
target site, as observed by NMR (23). We
estimate a dissociation constant of 25 ± 10 nM by photocleavage titration at
this doublet as a function of concentration based on curve fitting (see
Materials and Methods). Photocleavage quantum yields for these metal complexes
are quite low (19), yet it is clear that
binding saturation occurs well below 1 ![]() M.
The splitting of the 5' T into a double band is occasionally seen with
photocleavage on small oligonucleotides with
M.
The splitting of the 5' T into a double band is occasionally seen with
photocleavage on small oligonucleotides with ![]() -1-Rh(MGP)2phi5+
(35). No specific sites of photocleavage
activity in either oligonucleotide were observed for the metal complexes rac-Rh(phen)2phi3+
(data not shown) or
-1-Rh(MGP)2phi5+
(35). No specific sites of photocleavage
activity in either oligonucleotide were observed for the metal complexes rac-Rh(phen)2phi3+
(data not shown) or ![]() -2-Rh(MGP)2phi5+
(Figure S3 and S4, Supporting Information) at concentrations lower than 10-5
M.
-2-Rh(MGP)2phi5+
(Figure S3 and S4, Supporting Information) at concentrations lower than 10-5
M.
Isomer Specificity of Competition Between Metal Complexes and Target
Oligonucleotide. Competition for the target oligonucleotide between yAP-1
and either ![]() -1-Rh(MGP)2phi5+
or
-1-Rh(MGP)2phi5+
or ![]() -2-Rh(MGP)2phi5+
was examined by titrating with the metal complexes using gel retardation assays
(Figures 4 and 5A). As standard procedure for all gel shift assays in our
laboratory, reactions were loaded directed onto a running gel regardless of
incubation time. Protein binding thus measured decreased in a sigmoidal fashion
with increasing concentrations of the metal complex, albeit at different
concentrations for different species of metal complex. Curve fitting of the
ratios of the bound oligonucleotide to total oligonucleotide showed that the
concentration of
-2-Rh(MGP)2phi5+
was examined by titrating with the metal complexes using gel retardation assays
(Figures 4 and 5A). As standard procedure for all gel shift assays in our
laboratory, reactions were loaded directed onto a running gel regardless of
incubation time. Protein binding thus measured decreased in a sigmoidal fashion
with increasing concentrations of the metal complex, albeit at different
concentrations for different species of metal complex. Curve fitting of the
ratios of the bound oligonucleotide to total oligonucleotide showed that the
concentration of ![]() -1-Rh(MGP)2phi5+
required to reach half-dissociation of the protein-DNA sites was 118 nM (Figure
4). The complex
-1-Rh(MGP)2phi5+
required to reach half-dissociation of the protein-DNA sites was 118 nM (Figure
4). The complex ![]() -2-Rh(MGP)2phi5+,
in contrast, showed no disruption of the DNA-protein complex at concentrations
as high as 10
-2-Rh(MGP)2phi5+,
in contrast, showed no disruption of the DNA-protein complex at concentrations
as high as 10 ![]() M, but nonspecific
aggregation of the DNA-protein complexes was evident at higher rhodium
concentrations (Figure 5B). The aggregation did not occur until the metal
complex was over 250 times more concentrated than yAP-1 in the binding
reactions, and over 10 times higher in concentration than the nonspecific
carrier protein BSA. This behavior was not seen with
M, but nonspecific
aggregation of the DNA-protein complexes was evident at higher rhodium
concentrations (Figure 5B). The aggregation did not occur until the metal
complex was over 250 times more concentrated than yAP-1 in the binding
reactions, and over 10 times higher in concentration than the nonspecific
carrier protein BSA. This behavior was not seen with ![]() -1-Rh(MGP)2phi5+
at concentrations as high as 40
-1-Rh(MGP)2phi5+
at concentrations as high as 40 ![]() M. The
differential binding by
M. The
differential binding by ![]() -1-Rh(MGP)2phi5+
versus
-1-Rh(MGP)2phi5+
versus ![]() -2-Rh(MGP)2phi5+
clearly demonstrates isomer specificity in recognition by the metal complex
(Figure 6B). The geometric repositioning of the pendant guanidiniums [e.g.,
-2-Rh(MGP)2phi5+
clearly demonstrates isomer specificity in recognition by the metal complex
(Figure 6B). The geometric repositioning of the pendant guanidiniums [e.g., ![]() -1-Rh(MGP)2phi5+
versus
-1-Rh(MGP)2phi5+
versus ![]() -2-Rh(MGP)2phi5+]
increases by over 2 orders of magnitude the amount of metal complex needed to
disrupt competitively specific protein-DNA complexes.
-2-Rh(MGP)2phi5+]
increases by over 2 orders of magnitude the amount of metal complex needed to
disrupt competitively specific protein-DNA complexes.
| Figure 4 Competition between the protein yAP-1 and the metal complex |
|
 |
Figure 5 Control competition reactions for the protein yAP-1. (A) |
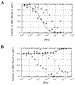 |
Figure 6 Comparison of fitted curves for competition between metal
complexes and yAP-1. Gel shift assays were quantitated in ImageQuant,
normalized, and then graphed and fitted in Kaleidograph. (A) Competition
between yAP-1 and |
Binding reactions with the parent metal complex rac-Rh(phen)2phi3+
were examined in similar competition experiments to explore the effect that a
relatively sequence neutral intercalating metal complex has on yAP-1 and
yAP-1/DNA complexes. rac-Rh(phen)2phi3+ does not
interfere with protein/DNA complexes directly, but competes at higher
concentration sterically for sites in the major groove of the oligonucleotide
(Figure 5C). Competition on both wild-type and target oligonucleotides had
half-site occupancy at 23 ![]() M metal complex
(Figure 6B). This value accurately reflects the lower overall binding affinity
that rac-Rh(phen)2phi3+, compared to
M metal complex
(Figure 6B). This value accurately reflects the lower overall binding affinity
that rac-Rh(phen)2phi3+, compared to ![]() -1-Rh(MGP)2phi5+,
displays for DNA. Indeed, previous work has estimated the dissociation constant
of this metal complex from DNA to be in the micromolar range (19),
and photocleavage experiments with the native and target oligonucleotides
described herein show no strong photocleavage bands at concentrations of metal
complex up to 20
-1-Rh(MGP)2phi5+,
displays for DNA. Indeed, previous work has estimated the dissociation constant
of this metal complex from DNA to be in the micromolar range (19),
and photocleavage experiments with the native and target oligonucleotides
described herein show no strong photocleavage bands at concentrations of metal
complex up to 20 ![]() M.
M.
Site-Specific Competition of yAP-1 and ![]() -1-Rh(MGP)2phi5+.
To demonstrate that successful competition between yAP-1 and
-1-Rh(MGP)2phi5+.
To demonstrate that successful competition between yAP-1 and ![]() -1-Rh(MGP)2phi5+
for a DNA-binding site is dependent on the presence of a target 5'-CATATG-3'
site, we performed identical competition experiments on both with the wild-type
ARE oligonucleotide and with the target oligonucleotide. In these experiments,
competition experiments using an oligonucleotide lacking a binding site for
-1-Rh(MGP)2phi5+
for a DNA-binding site is dependent on the presence of a target 5'-CATATG-3'
site, we performed identical competition experiments on both with the wild-type
ARE oligonucleotide and with the target oligonucleotide. In these experiments,
competition experiments using an oligonucleotide lacking a binding site for ![]() -1-Rh(MGP)2phi5+
(as shown by photocleavage, see Supporting Information) was compared to
experiments using the target oligonucleotide which contains the preferred
binding site 5'-CATATG-3'. If
-1-Rh(MGP)2phi5+
(as shown by photocleavage, see Supporting Information) was compared to
experiments using the target oligonucleotide which contains the preferred
binding site 5'-CATATG-3'. If ![]() -1-Rh(MGP)2phi5+
were nonspecifically interfering with the protein, identical behavior between
these two different protein-DNA complexes would be expected. However, as seen in
Figure 6A,
-1-Rh(MGP)2phi5+
were nonspecifically interfering with the protein, identical behavior between
these two different protein-DNA complexes would be expected. However, as seen in
Figure 6A, ![]() -1-Rh(MGP)2phi5+
disrupted 50% of the protein-target DNA complexes at a concentration of 120 nM,
whereas over 3
-1-Rh(MGP)2phi5+
disrupted 50% of the protein-target DNA complexes at a concentration of 120 nM,
whereas over 3 ![]() M rhodium was required to
compete similarly on the wild-type oligonucleotide. This result represents an
increase of 30 times the concentration of
M rhodium was required to
compete similarly on the wild-type oligonucleotide. This result represents an
increase of 30 times the concentration of ![]() -1-Rh(MGP)2phi5+
required to disrupt yAP-1-target oligonucleotide complexes (Figure 6A). In
effect, the removal of the binding site from the oligonucleotide increases the
concentration of metal complex required for successful competition with yAP-1 by
more than an order of magnitude.
-1-Rh(MGP)2phi5+
required to disrupt yAP-1-target oligonucleotide complexes (Figure 6A). In
effect, the removal of the binding site from the oligonucleotide increases the
concentration of metal complex required for successful competition with yAP-1 by
more than an order of magnitude.
| Figure 7 Schematic model of the competition between yAP-1 and |
Target Oligonucleotide Design. In general, the native ARE shows a
great deal of tolerance for variation of the nucleotides within its binding site
that are not closely contacted by the protein. However, attempts to introduce a
metal complex binding site that disrupts one or more of the closely contacted
base pairs resulted in a pronounced decrease in the binding affinity between
yAP-1 and the modified ARE (data not shown). Consistent with this notion, the
mutation of the binding site to include a metal complex binding region without
changing these essential base pairs, as shown in Figure 2, did not alter the
binding of yAP-1 to the oligonucleotide. Gel retardation assays showed that, at
an approximate 2:1 molar ratio in titrations, the binding of the target
oligonucleotide to the protein was quantitative at very low nanomolar
concentrations (typically 8-10 nM of protein). Photocleavage with ![]() -1-Rh(MGP)2phi5+
confirmed that the metal complex strongly recognized the target oligonucleotide
containing the three base pairs altered from the wild-type ARE.
-1-Rh(MGP)2phi5+
confirmed that the metal complex strongly recognized the target oligonucleotide
containing the three base pairs altered from the wild-type ARE.
Site Specificity of the Competition Reaction. The ability of a metal
complex to compete specifically at low concentrations with a transcription
factor for a promoter depends on the presence of an overlapping binding site for
the two species. Competition between yAP-1 and ![]() -1-Rh(MGP)2phi5+
for the target oligonucleotide showed disruption at concentrations more than an
order of magnitude lower than seen for competition between the same species for
the wild-type ARE. These controls show that the competitive disruption of the
binding of yAP-1 to the ARE depends on the specific DNA sequence and not on a
nonspecific interaction with yAP-1. Nonspecific interactions between
-1-Rh(MGP)2phi5+
for the target oligonucleotide showed disruption at concentrations more than an
order of magnitude lower than seen for competition between the same species for
the wild-type ARE. These controls show that the competitive disruption of the
binding of yAP-1 to the ARE depends on the specific DNA sequence and not on a
nonspecific interaction with yAP-1. Nonspecific interactions between ![]() -1-Rh(MGP)2phi5+
and the carrier BSA are also ruled out by the differential inhibition of yAP-1
binding to DNA by
-1-Rh(MGP)2phi5+
and the carrier BSA are also ruled out by the differential inhibition of yAP-1
binding to DNA by ![]() -1-Rh(MGP)2phi5+.
Interaction of the metal complex with BSA cannot account for the strongly
sequence-specific competition between the transcription factor and
-1-Rh(MGP)2phi5+.
Interaction of the metal complex with BSA cannot account for the strongly
sequence-specific competition between the transcription factor and ![]() -1-Rh(MGP)2phi5+
that occurs at concentrations of metal complex an order of magnitude below the
concentration of BSA (Figure 6A).
-1-Rh(MGP)2phi5+
that occurs at concentrations of metal complex an order of magnitude below the
concentration of BSA (Figure 6A).
There are pronounced differences in binding affinity for the two
oligonucleotides and the metal complex ![]() -1-Rh(MGP)2phi5+,
as seen both by photocleavage and by competition with yAP-1 for an
oligonucleotide. These differences show that the metal complex binding is
specific to the sequence introduced into the ARE and not a nonspecific metal
complex-protein interaction. The data furthermore show that the specific
inhibition of protein binding is dependent on the presence of 5'-CATATG-3'
within the promoter site.
-1-Rh(MGP)2phi5+,
as seen both by photocleavage and by competition with yAP-1 for an
oligonucleotide. These differences show that the metal complex binding is
specific to the sequence introduced into the ARE and not a nonspecific metal
complex-protein interaction. The data furthermore show that the specific
inhibition of protein binding is dependent on the presence of 5'-CATATG-3'
within the promoter site.
Isomer and Complex Specificity of the Competition Reaction. The
specificity in metal complex for ![]() -1-Rh(MGP)2phi5+
is demonstrated through the comparison between its competition reactions and
those of related complexes. Other metal complexes showed nonspecific
interference in the binding of yAP-1 to the target oligonucleotide. Moreover,
for other metal complexes the protein-promoter complex disruption was
insensitive to the sequence of the DNA used. Both wild-type ARE and the target
ARE show approximately the same susceptibility to disruption, as is expected for
a competition with metal that is not site specific. Furthermore, titrations
revealed that only concentrations of rac-Rh(phen)2phi3+
above 20
-1-Rh(MGP)2phi5+
is demonstrated through the comparison between its competition reactions and
those of related complexes. Other metal complexes showed nonspecific
interference in the binding of yAP-1 to the target oligonucleotide. Moreover,
for other metal complexes the protein-promoter complex disruption was
insensitive to the sequence of the DNA used. Both wild-type ARE and the target
ARE show approximately the same susceptibility to disruption, as is expected for
a competition with metal that is not site specific. Furthermore, titrations
revealed that only concentrations of rac-Rh(phen)2phi3+
above 20 ![]() M could disrupt DNA-protein
complexes. These concentrations are well over 2 orders of magnitude greater than
those seen for the specific competition of
M could disrupt DNA-protein
complexes. These concentrations are well over 2 orders of magnitude greater than
those seen for the specific competition of ![]() -1-Rh(MGP)2phi5+.
-1-Rh(MGP)2phi5+.
The symmetrical isomer ![]() -2-Rh(MGP)2phi5+
also showed no disruption of protein-DNA complexes up to 20
-2-Rh(MGP)2phi5+
also showed no disruption of protein-DNA complexes up to 20 ![]() M
either with the wild-type ARE or with the target ARE (Figure 6B). However, as
the concentration of the metal complex increased to over 250 times that of yAP-1
and over 10 times that of the carrier BSA, aggregation in the wells occurred.
Importantly, the only difference between this series of reactions and those
performed for
M
either with the wild-type ARE or with the target ARE (Figure 6B). However, as
the concentration of the metal complex increased to over 250 times that of yAP-1
and over 10 times that of the carrier BSA, aggregation in the wells occurred.
Importantly, the only difference between this series of reactions and those
performed for ![]() -1-Rh(MGP)2phi5+
was the orientation of the pendant guanidiniums. Positioning these arms so that
they no longer contact DNA removes all specificity for DNA recognition, as seen
previously (20). Inspection of the
geometry of the two isomeric species (see Figure 1) shows that
-1-Rh(MGP)2phi5+
was the orientation of the pendant guanidiniums. Positioning these arms so that
they no longer contact DNA removes all specificity for DNA recognition, as seen
previously (20). Inspection of the
geometry of the two isomeric species (see Figure 1) shows that ![]() -1-Rh(MGP)2phi5+
directs its guanidinium functional groups into the DNA major groove. In
contrast,
-1-Rh(MGP)2phi5+
directs its guanidinium functional groups into the DNA major groove. In
contrast, ![]() -2-Rh(MGP)2phi5+
has both guanidiniums directed away from the intercalating phi ligand. The
differences in orientation of hydrogen-bonding substituents between the two
complexes must be the source of the different biochemical interactions of these
metal complexes; indeed the overall charge on the complexes and thus their
electrostatic interaction with DNA is the same. The proximity of the two
guanidiniums in
-2-Rh(MGP)2phi5+
has both guanidiniums directed away from the intercalating phi ligand. The
differences in orientation of hydrogen-bonding substituents between the two
complexes must be the source of the different biochemical interactions of these
metal complexes; indeed the overall charge on the complexes and thus their
electrostatic interaction with DNA is the same. The proximity of the two
guanidiniums in ![]() -2-Rh(MGP)2phi5+
could be responsible for the increased inter-complex contacts between multiple
protein-DNA complexes we observe and, hence, cause a nonspecific aggregation not
dissimilar to that used in ammonium sulfate precipitations of susceptible
proteins in purification steps.
-2-Rh(MGP)2phi5+
could be responsible for the increased inter-complex contacts between multiple
protein-DNA complexes we observe and, hence, cause a nonspecific aggregation not
dissimilar to that used in ammonium sulfate precipitations of susceptible
proteins in purification steps.
Model for Competition. yAP-1 shares many features with other bZIP
transcription factors (36). Comparison of
our designed target site with the related oligonucleotide site in the crystal
structure of the GCN4 DNA-binding domain (37)
suggests that the presence of a metal complex in the location of our introduced
binding site would appear to block sterically one of the two dimer arms from
making contacts with half of the semi-symmetrical core of the binding domain.
All of the modifications to the ARE are located 5' to the pivotal G-C base pair,
and hence on only one of the two dimer binding regions. Although there still
exists some controversy regarding the dimerization and DNA binding pathway that
proteins follow in vivo, recent models have suggested that bZIP proteins
dimerize stepwise on some target sites of DNA (38,
39)
Nature offers many parallels to the type of competition ![]() -1-Rh(MGP)2phi5+
shows with yAP-1. By using direct competition at overlapping sites, often two
repressor proteins modulate gene expression. In much the same way, the
site-specific competition between metal complexes and transcription factors
might be exploited to modulate expression.
-1-Rh(MGP)2phi5+
shows with yAP-1. By using direct competition at overlapping sites, often two
repressor proteins modulate gene expression. In much the same way, the
site-specific competition between metal complexes and transcription factors
might be exploited to modulate expression.
A Promising New Use for Metallointercalators. Octahedral
metallointercalators would have to satisfy many criteria to be candidates for
possible therapeutic applications. Perhaps the first of these important criteria
would be the site-specific inhibition of a transcription factor binding to an
enhancer site. We have shown previously that metal complexes can inhibit
restriction endonucleases, and that metal complexes can mimic the binding
characteristics of transcription factors (13, 40)
We also thank Dr. Christine Rener for technical assistance.
Supporting data, which includes one two-part gel retardation experiment and three sequencing gels of photocleavage reactions and captions, is available. This material is available free of charge via the Internet at http://pubs.acs.org.
![]() We are grateful to the NIH
(GM33309 to J.K.B. and GM47381 to C.S.P.) for their financial support and for an
NRSA traineeship to D.T.O.
We are grateful to the NIH
(GM33309 to J.K.B. and GM47381 to C.S.P.) for their financial support and for an
NRSA traineeship to D.T.O.
* To whom correspondence should be addressed.
1. Ahmad, I., and Rao, D. N. (1996) Crit. Rev. Biochem.
Mol. Biol. 31, 361-380.
2. Donahue, B. A., Augot, M.,. Bellon, S. F., Treiber, D. K., Toney, J. H.,
Lippard, S. J., and Essigmann, J. M. (1990) Biochemistry 29, 5872-5880.
3. Pil, P. M., and Lippard, S. J., (1992) Science 256, 234-237.
4. Whitehead, J. P., and Lippard, S. J. (1996) Metal Ions Biol. Sys.
32, 687-726.
5. Tomasz, M., and Palom, Y. (1997) Pharm. Ther. 76,
73-87.
6. Poltev, V. I., Bruskov, V. I., Shulyupina, N. V., Rein, R., Shibata, M.,
Ornstein, R. L., and Miller, J. (1993) Mol. Biol. 27,
447-462.
7. Denison, C., and Kodadek, T. (1998) Chem. Biol. 5,
R129-R145.
8. Liu, C. Smith, B. M., Ajito, K., Komatsu, H., GomezPaloma, L., Li, T. H.,
Theodorakis, E. A., Nicolau, K. C., and Vogt, P. K. (1996) Proc. Natl.
Acad. Sci. U.S.A. 93, 940-944.
9. Nicolaou, K., Tsay, S. C., Suzukit, T., and Joyce, G. F. (1992)J.
Am. Chem. Soc. 114, 7555-7557.
10. Nicolaou, K., and Dai, W. (1991) Angew. Chem. 30,
1387-1416.
11. Wemmer, D. E., and Dervan, P. B. (1997) Curr. Opinion Struct.
Biol. 7, 355-361.
12. Gottesfeld, J., Neely, L., Trauger, J. W., Baird, E. E., and Dervan, P.
B. (1997) Nature 387, 202.
13. Terbrueggen, R. H., and Barton, J. K. (1995) Biochemistry 34,
8227-8234.
14. Johann, T. W., and Barton, J. K. (1996) Philos. Trans. R.
Soc. London A 354, 299-324.
15. Hudson, B. P., and Barton, J. K. (1998) J. Am. Chem.
Soc. 120, 6877-6888.
16. David, S. S., and Barton, J. K. (1993) J. Am. Chem.
Soc. 115, 2984-2985.
17. Shields, T. P., and Barton, J. K. (1995) Biochemistry 34,
15049-15056.
18. Krotz, A. H., Hudson, B. P., and Barton, J. K., (1993) J. Am.
Chem. Soc. 115, 12577-12578.
19. Sitlani, A., Long, E. C. Pyle, A. M., and Barton, J. K. (1992) J.
Am. Chem. Soc. 114, 2303-2312.
20. Terbrueggen, R. H., Johann, T. W., and Barton, J. K. (1998) Inorg.
Chem. 37, 6874-6883.
21. Campisi, D., Morii, T., and Barton, J. K. (1994) Biochemistry 33,
4130-4139.
22. Pabo, C. O., and Sauer, R. T. (1992) Annu. Rev. Biochem.
61, 1053.
23. Franklin, S. J., and Barton, J. K. (1998) Biochemistry 37,
16093-16105.
24. Kim, J. L., Nikolov, D. B., and Burley, S. K. (1993) Nature 365,
520-527.
25. Kim, Y. C., Geiger, J. H., Hahn, S., and Sigler, P. B. (1993) Nature
365, 512-520.
26. Oshea, E. K., Klemm, J. D., Kim, P. S., and Alber, T. (1991) Science
254, 539-544.
27. Moye-Rowley, W. S., Harshman, K. D., and Parker, C. S. (1988) Cold
Spring Harbor Symp. Quant. Biol. LIII, 711.
28. Moye-Rowley, W. S., Harshman, K. D., and Parker, C. (1989) Genes Dev.
3, 283-292;
29. Wemmie, J., Wu, A.-L., Harshman, K. D., Parker, C. S., and Moye-Rowley,
W. (1994) J. Biol. Chem. 269, 14690.
30. Harshman, K. D., Moye-Rowley, W. S., and Parker, C. (1988) Cell 53,
321-330.
31. Chow, C. S., and Barton, J. K. (1992) Methods Enzymol.
212, 219-242.
32. Kunkel, T. A. (1985) Proc. Natl. Acad. Sci.
U.S.A. 82, 488-492.
33. Singleton, S., and Dervan, P. B. (1992) J. Am. Chem.
Soc. 114, 6957.
34. LaValle, E. R., DiBlasio, E. A., Kovacic, S., Grant, K. L., Schendel, P.
F., and McCoy, J. M. (1993) BioTechnology 11, 187-193.
35. Johann, T. W. California Institute of Technology, Ph.D. Dissertation.
36. Vinson, C. R., Sigler, P. B., and McKnight, S. (1989) Science 246,
911.
37. Ellenberger, T. E., Brandl, C. J., Struhl, K., and Harrison, S. C. (1992)
Cell 71, 1223-1237.
38. Metallo, S. J., and Schepartz, A. (1997) Nat. Struct.
Biol. 4, 115-117.
39. Weiss, M. A. (1990) Biochemistry 29, 8020-8024.
40. Sitlani, A., Dupureur, C. M., and Barton, J. K. (1993) J. Am.
Chem. Soc. 115, 12589-12590.